Introduction
Antibodies against myelin oligodendrocyte glycoprotein (MOG-Abs) have been associated with non-multiple sclerosis inflammatory demyelinating central nervous system diseases, including acute disseminated encephalomyelitis (ADEM), encephalitis, optic neuritis, and myelitis [1]. Only a few cases of adults with MOG-Abs-associated disorders have been reported to manifest seizures and ADEM as the main clinical manifestations [2]. Because of this rarity, the clinical characteristics and treatment guidelines for the adult form of ADEM and the seizure phenotype in MOG-Abs-associated disorders have yet to be established.
Herein, we report a case of MOG-Abs-associated disorder in a patient who presented with status epilepticus (SE) as the index manifestation, followed by chronic epilepsy. We further discuss the management of chronic epilepsy in patients with MOG-Abs-associated disorders. Written informed consent was obtained for publication of this case report and accompanying images.
Case Report
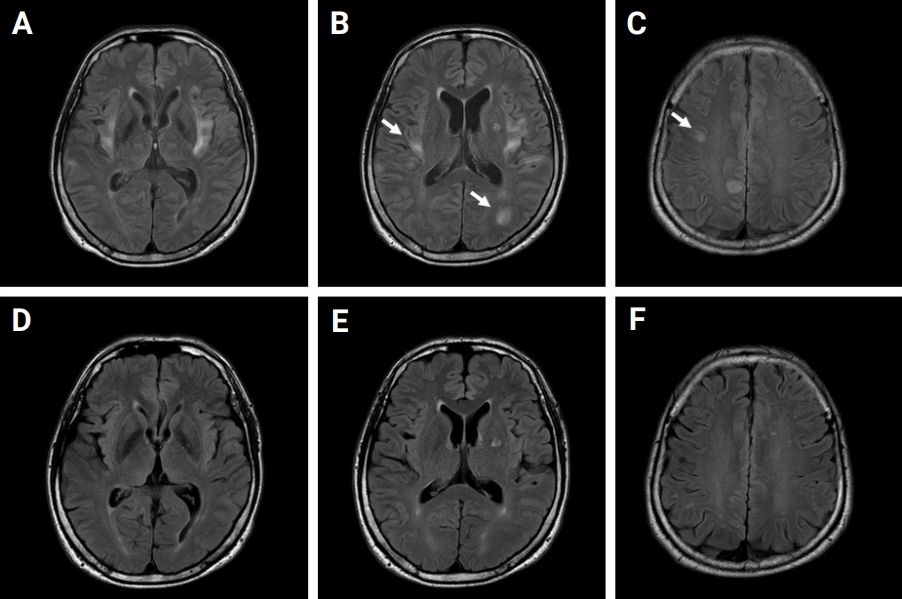
A 49-year-old male presented to the emergency department (ED) with first-ever seizure. He had no past medical history. In the ED, his body temperature was 37.9┬░C, and he manifested recurrent generalized tonic seizures without recovering baseline cognitive function between seizures. An intravenous (IV) dose of lorazepam followed by IV fosphenytoin and levetiracetam (LEV) failed to control recurrent convulsive seizures. The convulsive SE lasted for approximately 12 hours, and the patientŌĆÖs convulsive seizures finally subsided with thiopental therapy. A cerebrospinal fluid study was normal. IV acyclovir, methylprednisolone, and immunoglobulin were administered and maintained. After administration of IV methylprednisolone for 5 days, oral methylprednisolone was initiated and maintained at a dosage of 70 mg/day. Electroencephalogram (EEG) showed frequent spike and wave discharges arising from the left temporo-occipital lobe. Magnetic resonance imaging (MRI) with gadolinium enhancement of the brain demonstrated bilateral, asymmetric, multifocal, and hyperintense lesions involving mainly the juxtacortical subcortices and the bilateral insula without contrast enhancement or diffusion restriction (Figure 1AŌĆōC). The hyperintense centers of the lesions were surrounded by slightly less hyperintense areas with fuzzy margins (arrows in Figure 1).
An antibody study against MOG-Abs using cell-based assays with flow cytometry (CBA-FACS) yielded a positive test result, with an MOG-expressing cell binding ratio (MBR) of 12.29% (reference, positive of >9.94%). Other laboratory studies were conducted to diagnose connective tissue diseases, vasculitis, and any other systemic diseases, including tests for identification of antithyroid peroxidase Ab; thyrotropin receptor Ab; thyroglobulin Ab; lupus anticoagulant; antineutrophil cytoplasmic Ab; anti SS-related antigen B (anti La); human leukocyte antigens (HLA) B27, HLA B51, C3, and C4; double-stranded DNA Ab; antinuclear Ab; and CBA-FACS anti-aquaporin 4 Ab. These tests were all unremarkable. One month after admission to the hospital, the patientŌĆÖs mental status was fully recovered without any neurological sequelae. The patient was discharged from the hospital with maintenance medications of 300 mg/day of valproate (VPR) and 1,000 mg/day of LEV. After the patient was discharged from the hospital, methylprednisolone was gradually tapered over a period of 40 days and then discontinued, without seizure recurrence. The abnormal lesions previously detected on MRI were resolved in the follow-up brain MRI, suggesting demyelinating pathology rather than axonal injury (Figure 1DŌĆōF). However, MOG-Ab remained in the follow-up test (MBR, 10.55%).
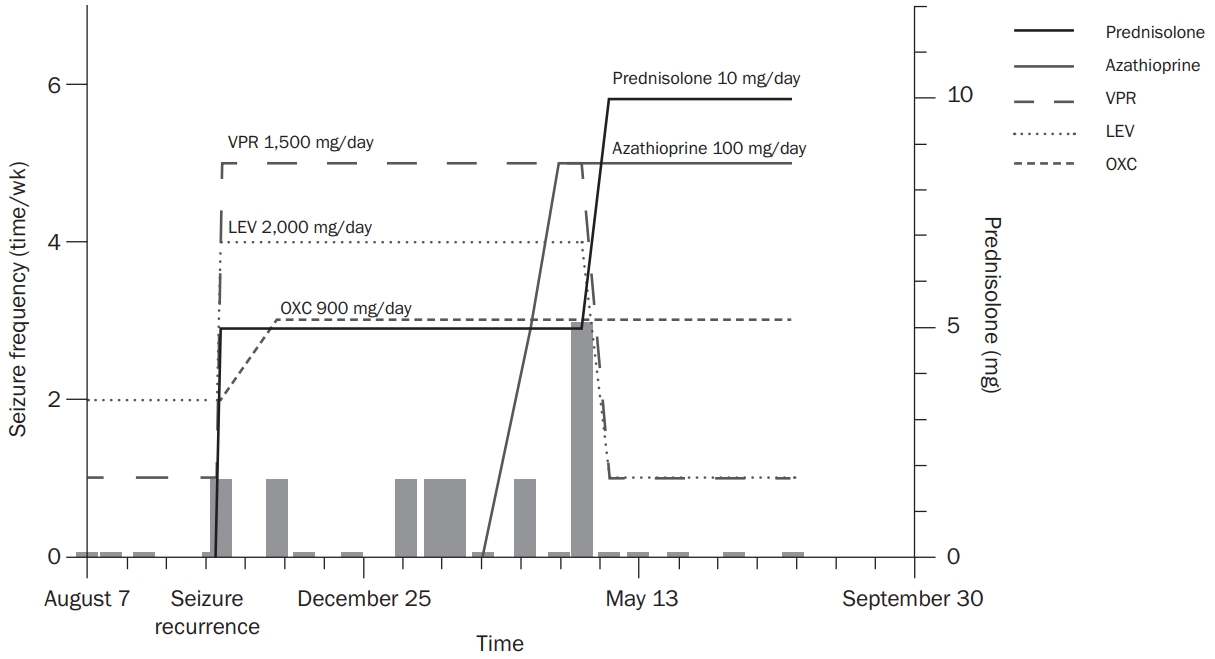
Thirty-six days after methylprednisolone was discontinued, the patient experienced a focal impaired awareness tonic seizure evolving to a bilateral tonic-clonic seizure. EEG revealed interictal spike and wave discharges from the left temporo-occipital lobe. The clinical and treatment courses regarding seizure frequency, antiseizure drugs (ASDs), and immunosuppressant agents are depicted in Figure 2. The dosages of ASDs before recurrence of seizure were 300 mg/day of VPR and 1,000 mg/day of LEV; after seizure recurrence, those were escalated to 1,500 mg/day of VPR and 2,000 mg/day of LEV, with newly added-on oxcarbazepine 900 mg/day.
The ASDs were not effective, and the frequency of seizures increased. The patientŌĆÖs drug-refractory epilepsy was eventually managed, and a seizure-free state was achieved by addition of azathioprine accompanied by titration of methylprednisolone from 5 to 10 mg/day. After effective response to immunotherapy, the doses of VPR and LEV were reduced to 300 and 500 mg/day, respectively.
Discussion
Analysis of the 23 most recently published studies in which the clinical and MRI characteristics of patients with MOG-Abs were evaluated clearly indicates age-dependent clinical features [1]. Young children most often present with ADEM with an opticospinal manifestation, though ADEM is very rare in adults [1]. None of the 26 adult patients with MOG-Abs in a case series manifested ADEM features on MRI [3]. In contrast, our patient showed multiple juxtacortical and cortical reversible brain lesions with blurred margins, compatible with the characteristic features of ADEM.
In 2017, an adult with focal and secondarily generalized seizures was reported and shown to be positive for MOG-Abs [2]. In that case, a relapse of optic neuritis occurred when methylprednisolone was tapered. Chronic epilepsy is very rare in MOG encephalitis. A retrospective study evaluating 61 MOG encephalomyelitis patients reported 13 patients (21.3%) with epileptic seizures, most of which were single, while SE was very rare (4%). Among the 13 patients with epileptic seizures, only two (15.4%) developed chronic epilepsy [4].
Although there is no established treatment consensus regarding the management of seizures in patients with MOG, a few studies reported that the seizures were responsive to steroids and/or immunosuppressants [1,2]. The present study provides evidence that patients with MOG-Abs and seizures are responsive to immunosuppressants and steroids based on a temporal relationship between ASDs, steroids, azathioprine, and seizure frequency. It has been reported that a subset of patients with medically intractable epilepsy have autoimmune epilepsy, and immunotherapy was very effective in these patients [5]. A trial of immunotherapy should be considered in patients with suspected autoimmune epilepsy.
In conclusion, we reported an adult patient who carried antibodies against MOG, presented with SE as the index event, and developed chronic epilepsy. The patient manifested ADEM-like features on MRI. Not sufficiently managed by ASDs, the chronic epilepsy was controlled by an immunosuppressant agent with a corticosteroid. Clinicians should use immunosuppressant agents as a first-line therapy for management of epilepsy in MOG-Ab-associated disorders.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print