Introduction
Myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD) encompasses a spectrum of autoimmune disorders with diverse clinical manifestations including myelitis, meningitis, encephalitis, optic neuritis, and acute disseminated encephalomyelitis. MOGAD rarely presents with unilateral cerebral cortical encephalitis (CCE), rendering diagnosis difficult in these cases. Since the first report of MOGAD presenting as a unilateral CCE [1], several cases of unilateral CCE have been reported [2]. Based on previous reports, patients with unilateral CCE frequently present with seizures, followed by headache, fever, and cortical symptoms [2] with characteristic magnetic resonance imaging (MRI) findings of unilateral cortical hyperintensities [3].
MOGAD is frequently associated with comorbid autoimmune diseases. In a previous study, 4% to 17% of patients with MOGAD reportedly exhibited comorbid autoimmune diseases such as thyroid disease or inflammatory bowel disease [4]. Therefore, evaluation for coexisting autoimmune diseases in patients with MOGAD is important.
Herein, we report a case of MOGAD presenting as unilateral CCE, which to the best of our knowledge, has not been reported in Korea. Our patient presented with additional systemic symptoms and was diagnosed with comorbid ulcerative colitis (UC).
We believe this case introduces unilateral CCE as a rare presentation of MOGAD and highlights the need to assess for comorbid autoimmune diseases in patients with MOGAD.
This study was approved by the Institutional Review Board of the Dongguk University Ilsan Hospital (No. 2024-03-012), and the exemption for written informed consent has been approved for publication of this report and accompanying images.
Case Report
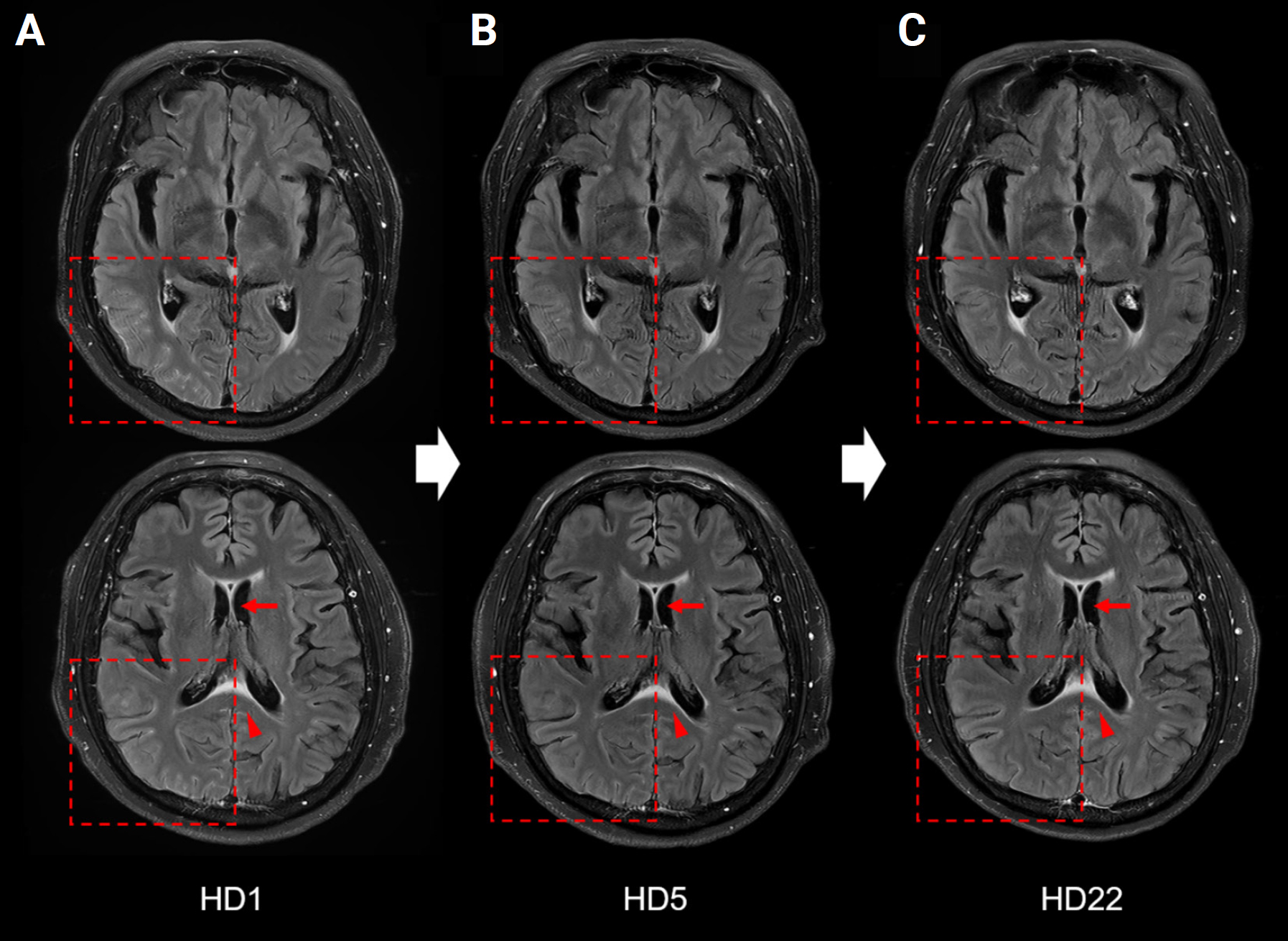
A 60-year-old female was admitted to our hospital with an acute-onset headache. The pulsatile headache in the right frontal area was accompanied by nausea, vomiting, diarrhea, and abdominal pain. She had a low-grade fever with a temperature of 38.1 ºC. A physical examination revealed abdominal tenderness, particularly on the right side and meningeal irritation signs were not present. She was alert and oriented to time, place, and person. She had left-sided homonymous hemianopsia and intermittent visual hallucinations lasting approximately 2 to 3 minutes. She described the visual hallucinations as flickering red-circular spots in the left visual field. During the visual hallucination, she showed a dull response to external stimuli. Laboratory analysis revealed an absence of leukocytosis; however, the C-reactive protein level was elevated to 1.48 mg/L. Her cerebrospinal fluid (CSF) test was normal (opening pressure, 12.6 cmH2O; white blood cell count, 2/mm3; protein, 45.3 mg/dL; glucose, 94 mg/dL) and showed no evidence of either bacterial or viral infection. Brain MRI showed focal high-signal intensity in lesions in the right parietal cortex, lower corpus callosum, and fornix, with diffuse sulcal enhancement along the right temporo-parieto-occipital sulci (Figure 1A). Electroencephalography (EEG) that was performed 6 hours after the onset of symptoms showed slow (7 Hz) waves in the right parieto-occipital lobe (Figure 2A). Considering the intermittent visual hallucinations and EEG findings, the patient was diagnosed with occipital lobe epilepsy and administered an antiepileptic drug, levetiracetam 250 mg, twice a day. Subsequently, her headache, hemianopsia, and visual hallucinations improved and partial improvement was observed on both brain MRI and EEG (Figures 1 and 2).
However, due to persisting abdominal symptoms, a sigmoidoscopy was performed and showed extensive erosion and ulceration in the colon. Based on endoscopic findings and laboratory results, UC was diagnosed. She was treated with 100 mg intravenous hydrocortisone every 8 hours for 3 days and switched to oral prednisolone for 5 weeks. After the steroid treatment, her abdominal symptoms improved. Anti-myelin oligodendrocyte glycoprotein (MOG) antibodies were detected in the blood using a fluorescence-activated cell sorting live cell assay. However, anti-MOG antibodies were not found in CSF due to a lack of laboratory facilities in our hospital. Other autoantibodies such as anti-aquaporin-4 antibody, N-methyl-ᴅ-aspartate receptor, leucine-rich glioma-inactivated 1 immunoglobulin G (IgG), and contactin-associated protein 2 IgG, were all negative. Ultimately, the patient was diagnosed with MOGAD and concurrent UC. Because she was treated with steroids for UC and her symptoms improved, no further immunosuppressive therapy was administered (Figure 3).
Discussion
Anti-MOG-associated encephalitis typically involves the supratentorial deep white matter, cortical gray juxtacortical white matter, corpus callosum, pons, cerebellum, midbrain, and medulla [5]. However, patients with anti-MOG antibodies may show atypical presentations such as unilateral CCE [5]. Because unilateral CCE can only be suspected on detailed neurological examination along with MRI testing, diagnosing anti-MOG-associated unilateral CCE is difficult for physicians. Furthermore, various neurologic disorders such as viral encephalitis, Rasmussen encephalitis (RE), Creutzfeldt-Jakob disease (CJD), and autoimmune encephalitis can also cause unilateral CCE [5]. Therefore, physicians should include this rare but potentially curable disease in their differential diagnosis when assessing unilateral CCE.
In this case report, the patient presented with symptoms including headache, fever, and seizures and showed hyperintense lesions in the cerebral cortex on brain MRI and a positive result for anti-MOG antibodies in the serum. Other potential causes such as RE, CJD, infection, and other autoimmune encephalitis were excluded based on clinical, radiological, and laboratory results.
The patient was diagnosed with comorbid UC. Reportedly, up to 10% of patients with MOGAD have comorbid autoimmune diseases [6]. Salama et al. [4] reviewed 23 patients with MOGAD and approximately one-quarter of the patients had a history of other autoimmune disorders such as psoriasis, thyroid disease, and UC. Almost one-third of those patients had a family history of autoimmune disorders. Therefore, we suggest that when a patient with MOGAD presents with systemic symptoms involving the skin and gastrointestinal tract, physicians should consider the possibility of comorbid autoimmune disorders.
In conclusion, the present case provides an example of unilateral CCE as a rare presentation of MOGAD in Korea and highlights the importance of assessing for comorbid autoimmune disorders in patients with MOGAD.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print