Introduction
N-methyl D-aspartate receptor (NMDAR) encephalitis is the most common antibody-mediated autoimmune encephalitis that frequently affects young females [1-4]. Ovarian teratoma, herpes simplex encephalitis, or other autoimmune diseases such as neuromyelitis optica spectrum disorder are known to trigger the production of immunoglobulin G1 (IgG1) and promote autoantibody binding to the NR1 subunit of NMDAR, resulting in the hypofunction of NMDA signals in neuronal synapses [5-8].
NMDAR encephalitis is a well-characterized clinical syndrome [9,10]. After the prodromal manifestation of fever and headache, typical symptoms occur that involve the limbic system, including memory dysfunction, psychiatric symptoms, language disturbance, and seizures. Once developed, encephalitis tends to rapidly deteriorate neural function, with patients exhibiting intractable seizures, dyskinesia, dysautonomia, central hypoventilation, and coma [4,11]. Although NMDAR encephalitis has a monophasic course with a near-complete recovery of symptoms in most cases, long-term sequelae such as executive dysfunction, psychomotor slowing, disinhibition, and disturbed sleep remain in some severe and intractable cases [9-14]. However, the mechanism for the anti-NMDAR autoantibody-related development of these characteristic clinical features and patterns of progression has not been fully addressed.
Here, we briefly introduce several pathomechanistic hypotheses that explain how NMDAR hypofunction causes the typical symptoms and prognosis of NMDAR encephalitis.
NMDAR and Its Function
NMDAR is one of the three major glutamate-gated cation channels, along with α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor and kainate receptor. NMDAR is a tetramer comprised of two NR1 and two NR2 or NR3 subunits [15]. Alternative splicing of the GRIN1 gene results in eight isoforms of the NR1 subunit; NR2 and NR3 are also classified into four (NR2A–NR2D) and two (NR3A–NR3B) isoforms, respectively. The combination of these isoforms makes up various subtypes of NMDARs with different spatial distributions and molecular functions [15,16].
Binding of glycine/D-serine to the NR1 subunit or binding of glutamate/NMDA to the NR2/3 subunit activates NMDARs and results in the flow of Na+ and Ca2+ into neurons and K+ out of neurons. By means of Ca2+ influx, NMDAR mediates various intra-neuronal signaling pathways [6,9,17,18]. NMDAR is also involved in synaptic plasticity, which is crucial for learning and memory, and a certain level of NMDA signaling is also critical for maintaining neuronal survival [19-24].
Anti-NMDAR Autoantibody Mechanism of Action
The main mechanism of the NMDAR autoantibody is the anti-NMDAR autoantibody-provoked cross-linking of NMDARs which inhibits NMDAR’s interaction with ephrin B2, a synaptic protein that stabilizes NMDAR for its placement in the synaptic structure [5,6,8,9,18,25,26]. Consequently, anti-NMDAR autoantibodies cause the NMDAR hypofunction by removing NMDAR from the synaptic surface by receptor internalization or by repositioning to the extrasynaptic surface [5,6,8,9,18,25]. Binding of IgG1 autoantibodies to NMDARs induces the transient NMDAR-mediated hyperactivation of neurons, which manifests as seizure, dyskinesia, and dystonia, and provokes excitotoxic damage to neurons. Additionally, IgG1 autoantibodies have a high affinity for the Fcγ receptor, which enables the complement activation and provocation of T-cell mediated autoimmunity, resulting in permanent damage to neurons [6]. This mechanism might be an explanation for long-term neurological deficit after severe and refractory NMDAR encephalitis associated with diffuse cerebral atrophy and progressive cerebellar atrophy [9,10,12,13].
Pathomechanism of Rapidly Progressive Limbic Dysfunction
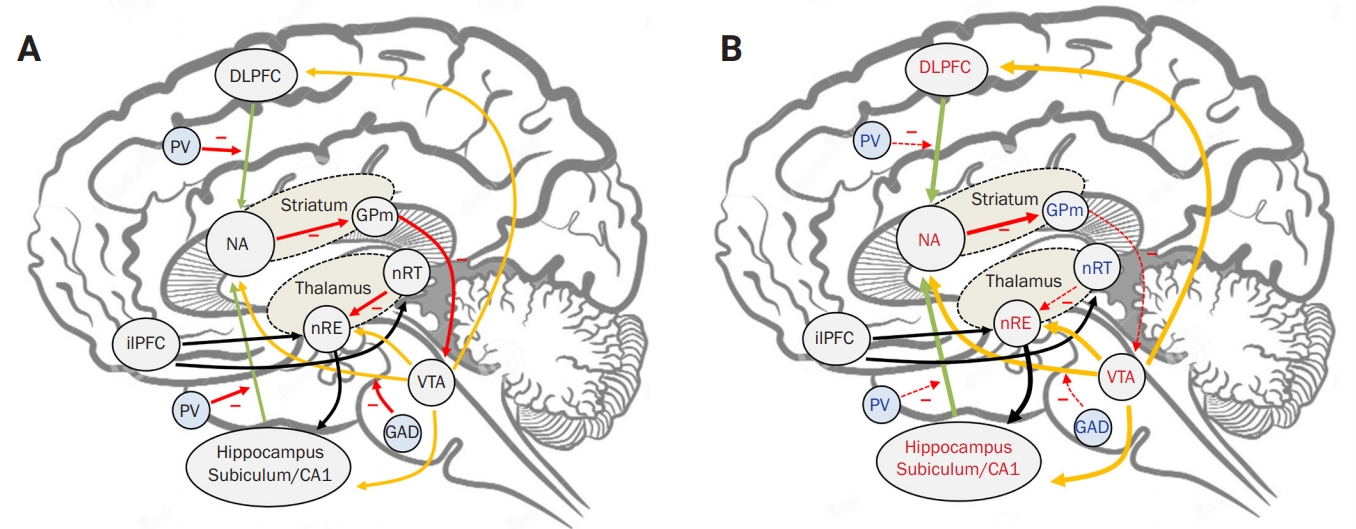
Given that the typical symptoms involving the limbic system in NMDAR encephalitis resemble the core symptoms of schizophrenia, the NMDA-mediated pathomechanism of schizophrenia might be successfully applied to understand the symptomatologic mechanism of NMDAR encephalitis [9,10,27]. In the schizophrenia model, parvalbumin or glutamate decarboxylase (GAD) positive inhibitory interneurons activated by NMDAR have a key mechanistic role. Parvalbumin-positive interneurons exert gamma-aminobutyric acid (GABA)-dependent inhibition on glutamatergic pyramidal neurons in the dorsolateral prefrontal cortex (DLPFC) and the subiculum of the hippocampus, which excites the nucleus accumbens (NA) of the striatum. NA exerts inhibitory regulation on the globus pallidus medialis (GPm), and the GPm also inhibits the ventral tegmental area (VTA) in the midbrain. Excitatory dopaminergic neurons in the VTA stimulate the DLPFC, NA, and hippocampus while receiving inhibitory regulation from GAD-positive interneurons [9,10,27].
Taken together, the DLPFC/subiculum, NA, and VTA form a closed loop of simulative interaction, and NMDAR-containing interneurons have a critical role as a restrainer of this positive feedback system [9,10,27]. Hypofunction of NMDAR in this system induces hyperactivation of the NA and VTA by disinhibition, resulting in dysregulated excitation of the DLPFC and subiculum (Figure 1). Given that DLPFC is the fundamental structure responsible for executive function and the integration of other cognitive functions and the subiculum is the main output structure of the hippocampus, dysregulated hyperactivation of these structures explains memory, language, and other cognitive dysfunctions [28-30]. Additionally, the VTA exerts widespread dopaminergic stimulatory projections to the cerebral cortex and limbic structures, which are called mesocortical and mesolimbic pathways, respectively [31]. Therefore, unregulated hyperfunction of the VTA might be responsible for psychiatric symptoms along with working memory deficit and global cognitive impairment in NMDAR encephalitis [27]. However, in contrast to NMDAR encephalitis, schizophrenia is a disorder with a high genetic predisposition, and its symptoms contain both positive and negative symptoms of dopaminergic dysregulation. Additionally, as well as NMDAR hypofunction, the disrupted balance between NMDA and AMPA receptors also has a significant role in the pathomechanism of schizophrenia [32]. The differences between these two diseases should be investigated to further elucidate the pathomechanism of NMDAR encephalitis.
The unregulated auto-activation of the DLPFC/subiculum–NA–VTA system also explains the rapidly progressing features of disease at the acute stages. Due to the loss of the NMDAR restrainer, the stimulatory interaction among DLPFC/subiculum, NA, and VTA strengthens by itself via this positive feedback loop, manifesting as an accelerated disease progression to the neurological nadir [9-11].
Extreme Delta Brush: Role of the Thalamus
Although not yet elucidated due to the high complexity of its involved circuits, thalamic structures might be critically involved in the pathogenesis of extreme delta brush, the core features of NMDAR encephalitis [33]. This hypothesis is supported by descriptions of abnormally enhanced delta and beta-gamma activities in both schizophrenia and NDMAR encephalitis [33-36] and research demonstrating that the thalamus is the main generator of the brain activities of those frequency bands during wakefulness [37-43].
The first pathologically important thalamic structure is the nucleus reuniens (nRE). The nRE is located in the medial thalamus and exerts excitatory signaling on the cornu ammonis 1 and the subiculum of the hippocampus [43,44]. Thus, the nRE facilitates activation of VTA via the subiculum–NA–VTA pathway [43,44]. As activation of dopaminergic cells in the VTA results in the reciprocal enhancement of thalamic bursting, the nRE–hippocampus–VTA constitutes another loop of simulative interaction.
The second thalamic structure of interest is the nucleus reticularis thalami (nRT) of the dorsal thalamus, a crucial structure that regulates cortical wakefulness [37-39]. The nRT is composed of parvalbumin-positive GABAergic neurons activated by NMDARs. GABAergic neurons in the nRT exert a strong inhibitory regulation on thalamocortical neurons, and their activation results in strong cortical deactivation [40]. The inhibitory effect of nRT on thalamocortical neurons might is justified given that the nRT is the main inhibitor of the nRE, and thereby regulates general activation of the cortex via suppressing the nRE–hippocampus–VTA loop [43,45]. Due to these network properties, the nRT is regarded as the thalamic pacemaker of the cortex [40-42]. The infralimbic subdivision of the medial prefrontal cortex also contributes to the balance between nRE-mediated cortical activation and nRT-mediated depression by exerting stimulatory signals on both the nRT and nRE [43,45].
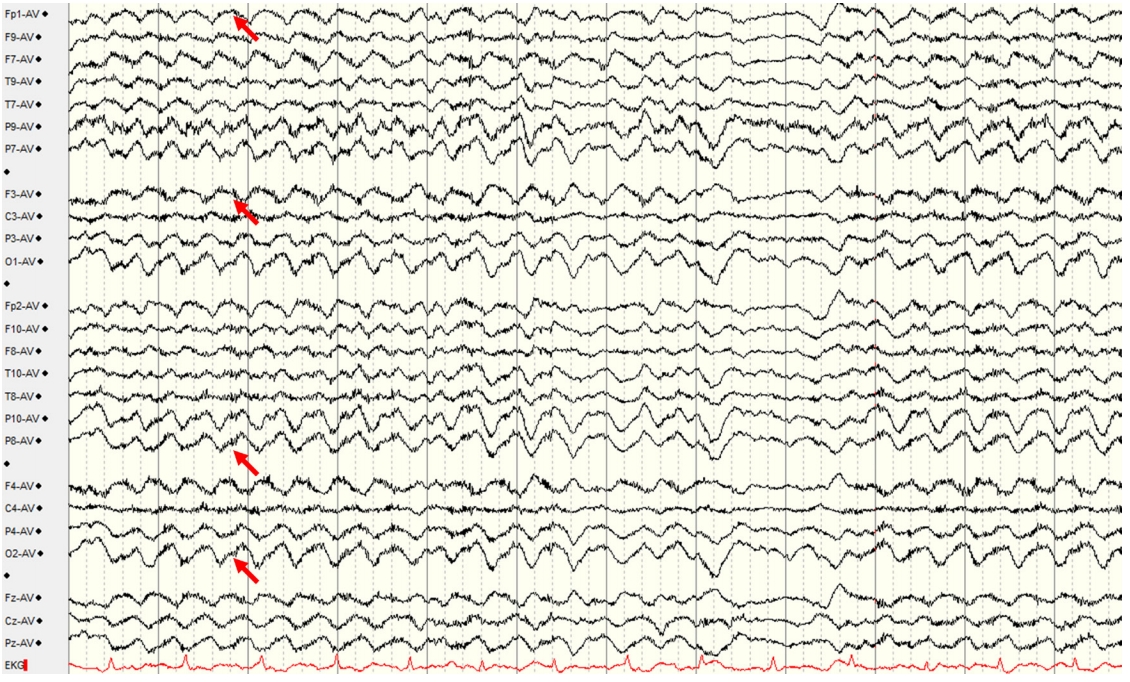
As the nRT’s GABAergic inhibition on the nRE is mediated by NMDAR, the nRE is the most highly activated thalamic structure by NMDAR antagonism [34,36]. Loss of NMDAR-mediated inhibition from nRT can enhance the burst activation of nRE by the nRE–hippocampus–VTA positive feedback loop, resulting in the formation of a rhythmic, synchronized activity of nRE (Figure 1) [34,36]. Accordingly, the cortex might be synchronized with the rhythmic activity of nRE via thalamocortical connections and projections from the VTA [34,36,40-42]. Synchronized low-frequency rhythmic activity in the cortex might appear as a frontal dominant rhythmic delta activity, a core electroencephalography feature of severe NMDAR encephalitis [33]. Fast activities with beta to gamma frequency bands superimposed with rhythmic delta activities, constituting another major feature of extreme delta brush, might represent the increased rhythmic cortical activity (Figure 2) [33,35,46,47]. This hypothesis is consistent with a recent study which demonstrated that pharmacological inhibition of the nRT resulted in the marked increment of cortical, delta, and gamma activity during wakefulness [36].
Pathomechanism of Consciousness Decrement, Seizure, Dyskinesia, and Psychosis
The thalamic hypothesis might also be applied to explain other major symptoms of NMDAR encephalitis. Multiple neurotransmitter systems involving the brainstem, hypothalamus, thalamus, and basal forebrain form a complex interaction to regulate cortical wakefulness. Among them, the nRT is a key structure for maintaining wakefulness [38,42]. Although activation of the nRT induces local suppression of cortical activity, global hypofunction of the NMDAR-dependent nRT provokes an unregulated burst activation of the nRE–hippocampus–VTA loop [38,42]. This results in the predominate delta rhythm throughout the cortex, which may critically contribute to the decrement of consciousness [38,41,42].
The major pathomechanism of intractable seizure development in NMDAR encephalitis is the shutdown of NMDR-dependent, parvalbumin- or GAD-positive inhibitory interneurons in the cortex and the thalamus [6,9,27]. Synchronized rhythmic cortical activation resulting from unregulated activation of the DLPFC/subiculum–NA–VTA and nRE–hippocampus‒VTA positive feedback loops maximizes the susceptibility to seizure development [9,33,48] and can fulfill the definition of a seizure in itself [49]. Additionally, binding of autoantibodies to NMDAR might provoke a transient activation of NMDAR, which provokes the flow of Na+ and Ca2+ into neurons and K+ out of neurons [6,17,50]. This antibody binding-mediated neuronal excitation might partially contribute to the development of seizures at very acute stages of disease.
Dyskinesia is a frequently encountered symptom of NMDAR encephalitis, especially among severe cases [4]. Although the underlying mechanisms of dyskinesia are heterogeneous and largely unrevealed, a recent study using a rat model of NMDAR encephalitis suggested that levodopa-induced dyskinesia is strongly associated with predominant gamma-band oscillation in the primary motor cortex [51,52]. The use of a dopamine antagonist suppressed this gamma-band oscillation along and improved dyskinesia [51,52]. The authors suggested that prominent gamma oscillation represents VPA-mediated primary motor cortex hyperactivation, which might be responsible for the development of dyskinesia [51,52]. The hypothesis of dopamine-mediated cortical hyperactivation can also explain the pathomechanism of dyskinesia in NMDAR encephalitis, as the unregulated activation of both the DLPFC/subiculum–NA–VTA and the nRE–hippocampus–VTA results in enhancement of dopaminergic cortex activation by VTA [35,46].
In the NMDA model of schizophrenia, it is speculated that individuals susceptible to the disease have partially compromised NMDAR function [10,34,44]. A triggering event such as acute stress stimulates dopaminergic activity, which can turn on the DLPFC/subiculum–NA–VTA and the nRE–hippocampus–VTA systems to an auto-reactivated status by means of a positive feedback loop [9,10,27,43,44]. Once activated, positive feedback loops contribute to the sustained activation of the VTA, resulting in the persisting symptoms of schizophrenia even after the initial trigger is removed [10,27,34,44]. This hypothesis can be applied to understand psychosis in NMDAR encephalitis, which is prominent at the initial disease presentation and at the recovering stages [9,10,14,53]. At those disease stages, the level and activity of anti-NMDAR antibody might not accumulate to reach a fulminant shut down of NMDAR function, but sufficiently diminish the NMDAR-mediated inhibition of the positive feedback systems to be activated by a certain trigger, resulting in delirium, disinhibition, and aggressive and compulsive behaviors [14,53].
Cerebellar Atrophy in NMDAR Encephalitis and Its Implication on Long-term Sequelae
NMDAR encephalitis exhibits two types of brain atrophy; diffuse cerebral atrophy and cerebellar atrophy [54-57]. Although diffuse cerebral atrophy is reversible and has an unclear association with long-term outcomes, cerebellar atrophy is progressive, irreversible, and associated with poor long-term clinical outcomes [54-57]. In our long-term cohort of NMDAR encephalitis patients with recurrent magnetic resonance imaging evaluations, we observed a significant reduction in both cerebellum and cerebrum volume, although the degree was higher in the cerebellum. Cerebellar atrophy was identified in 23.3% of the total study population and followed a progressive course once developed. The degree of cerebellar volume reduction was correlated with poorer scores of long-term global neurological outcomes and major functional domains such as memory, language, and psychiatric symptoms, which indicate that the degree of cerebellar atrophy is associated with the extent of permanent neurological deficits. The degree of cerebellar volume reduction was also associated with an increased cumulative disease burden (unpublished data).
High degrees of cerebellar atrophy suggest that the cerebellum might be especially vulnerable to brain atrophy in NMDAR encephalitis [19,58]. Previous studies demonstrated that a physiological level of NMDARs is required to maintain neuronal survival due to NMDAR-dependent Ca2+-mediated signaling. The cAMP response element-binding protein, extracellular signal-regulated kinase, calcium/calmodulin-dependent kinase II, and nuclear factor kappa-B pathways are required for the downstream activation of brain-derived neurotrophic factor [19-24]. Therefore, autoantibody-mediated depletion of NMDAR below a certain level might provoke neuronal degeneration. Considering that cerebellar granule cells, comprising more than 50% of the neurons in the brain, are highly abundant with NMDAR, the cerebellum might be the most highly affected by NMDAR depletion [59].
The progressive features of cerebellar atrophy imply that a long-term mechanism of neuronal degeneration might persist. A previous study demonstrated chronic persistence of autoantibodies in cerebrospinal fluid, even in patients with favorable recovery, and a slighter decrease of autoantibody was correlated with worse outcomes [60]. This finding suggests that the degree of cerebellar atrophy represents the cumulative effect of long-term NMDAR depletion by persisting autoantibodies [6,9,18]. In this regard, cerebellar volume reduction might be utilized as a surrogate marker for the cumulative burden of disease, which is also associated with long-term outcomes. The association of cerebellar volume reduction with functional outcomes might also be explained by the complex interconnection of the cerebellum with the limbic areas and neocortex [59,61-63].
Therapeutic Implications
Accelerated activation of the DLPFC/subiculum–NA–VTA and the nRE–hippocampus–VTA systems via positive feedback explains intractability at the fulminant status and the rapid progression of disease to the neurological nadir at acute stages of disease. In this regard, the use of combination immunotherapy at early disease stages might be rationalized, especially in cases with high clinical severity or rapid progression, to restore the NMDAR-mediated regulatory system and effectively interrupt the self-accelerated activation of these systems [11]. Furthermore, given that unregulated activation of the VTA-mediated dopaminergic pathway is critically involved in consciousness decrement, seizure, dyskinesia, and psychosis in NMDAR encephalitis, drugs affecting the dopaminergic system should be used with special attention to their adverse effects.
The irreversible and progressive feature of cerebellar atrophy, of which its degree is correlated with long-term neurological deficits, also supports the benefit of the early introduction of combination immunotherapy over the conventional stepwise use of immunotherapy, as it promotes early control of the disease and therefore can minimize the cumulative disease burden [11,64,65]. Additionally, in patients with persisting neurological symptoms in the chronic phase, adjuvant immunotherapy might be rationalized to interrupt the ongoing disease activity, halt the progression of cerebellar atrophy, and prevent long-term sequelae [11,64-68].
Conclusion
The main molecular mechanism of NMDAR encephalitis is autoantibody-mediated NMDAR hypofunction in the neuronal synapse. This results in the suppression of the NMDAR-dependent GABAergic interneurons, provoking accelerated activation of the DLPFC/subiculum–NA–VTA and the nRE–hippocampus–VTA systems via positive feedback which explains the rapidly deteriorating clinical course to the neurological nadir. Dysregulated activation of the VTA and cortex via those positive feedback loops might explain the well-characterized clinical syndrome of NMDAR encephalitis that includes limbic system dysfunction, intractable seizures, dyskinesia, coma, and the characteristic extreme delta brush. Progressive cerebellar atrophy is identified in severe and intractable cases, and its degree is correlated with the cumulative disease burden and worse outcomes, which might be explained by the NMDAR-dependent pathways for maintaining the cerebellar granule cell survival. Those pathomechanistic hypotheses of NMDAR encephalitis support the rationale for the early introduction of combination immunotherapy and the use of adjuvant immunotherapy in patients with persisting symptoms in chronic phases.
It should be noted that the symptomatologic pathomechanisms introduced here are hypothetical. Given the abundant distribution of NMDAR throughout the brain and the high complexity of functional interactions among the structures addressed in this review, further investigations should be aimed to elucidate the exact pathomechanisms of NMDAR encephalitis symptoms by using in vivo models or by functional or perfusion imaging analyses matched to quantitative electroencephalography data.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print