Herpes simplex encephalitis initially presenting without fever or cerebrospinal fluid pleocytosis and with typical neuroimaging findings: a case report
Article information

Abstract
Herpes simplex encephalitis (HSE) is a common viral encephalitis that can be fatal if not adequately treated. Fever, cerebrospinal fluid (CSF) pleocytosis, and typical neuroimaging findings are commonly observed in HSE cases. We encountered a patient with HSE who did not exhibit these classic clinical features. A 63-year-old male presented with his first-ever seizure. Fever did not develop until the fourth day of admission, and neither neuroimaging nor CSF analysis revealed abnormalities. Under suspicion of autoimmune encephalitis, methylprednisolone was administered. Subsequently, when the patient developed fever, a follow-up neuroimaging study was performed and revealed abnormalities consistent with HSE. The patient was promptly treated with acyclovir, which led to a full recovery. Diagnosing HSE in patients who present without fever or CSF pleocytosis and with typical neuroimaging findings poses a challenge. Therefore, prior to initiating immunosuppressive treatment, it is crucial to closely observe patients and to conduct follow-up tests, including neuroimaging and CSF analysis.
Introduction
Herpes simplex encephalitis (HSE), which is caused by the herpes simplex virus (HSV), is the most common viral encephalitis among adults [1] and commonly presents with high fever, altered mentation, and seizures [2]. Before the introduction of acyclovir, the mortality rate of HSE was as high as 70% to 80%. Acyclovir has reduced the mortality rate to 20%–30% [3].
HSE can be diagnosed through clinical history, neurological examination, cerebrospinal fluid (CSF) examination, electroencephalography (EEG), and neuroimaging, such as magnetic resonance imaging (MRI). The presence of either fever, CSF pleocytosis, or both is almost always seen in HSE [4]. In their absence, early diagnosis of HSE is difficult. We encountered a patient with HSE who presented without fever or CSF pleocytosis and report the clinical course and significance of this case.
Case Report
A 63-year-old male visited the emergency department of our hospital because he experienced a seizure for the first time in his life. A generalized tonic-clonic seizure that lasted for approximately 2 minutes and was accompanied by tongue biting was witnessed by the patient’s son. He reported a moderate-to-severe headache with nausea and vomiting 2 days prior to the seizure. The headache was throbbing in nature and was initially located in the temporal and frontal areas before spreading to the entire head. The symptoms were aggravated by overeating. He and his family denied any history of painkillers for the headache.
At the emergency room, the patient was mildly drowsy but could comply with the commands of the medical personnel. His vital signs were stable, and his body temperature was 36.7 °C. His orientation was relatively intact, although some errors were noted in the time orientation task (3 of 5) of the Korean Mini-Mental Status Examination (K-MMSE). However, his orientation regarding place and person was intact. The patient’s Glasgow Coma Scale score was E3V5M6. Focal neurological abnormalities (including neck stiffness) were not observed upon examination. He was admitted to the neurological intensive care unit (ICU) for close observation of mild drowsiness. Routine blood tests revealed no abnormalities except mild elevation of creatine phosphokinase (216 U/L; normal value, 46–171 U/L). MRI, which included diffusion-weighted imaging, showed no abnormalities (Figure 1). CSF examination revealed a white blood cell count, glucose level, and protein level of 4/μL (0–5/μL), 72 mg/dL (50–80 mg/dL), and 42.7 mg/dL (15–45 mg/dL), respectively. To prevent seizure recurrence, valproic acid (300 mg three times a day) was administered.
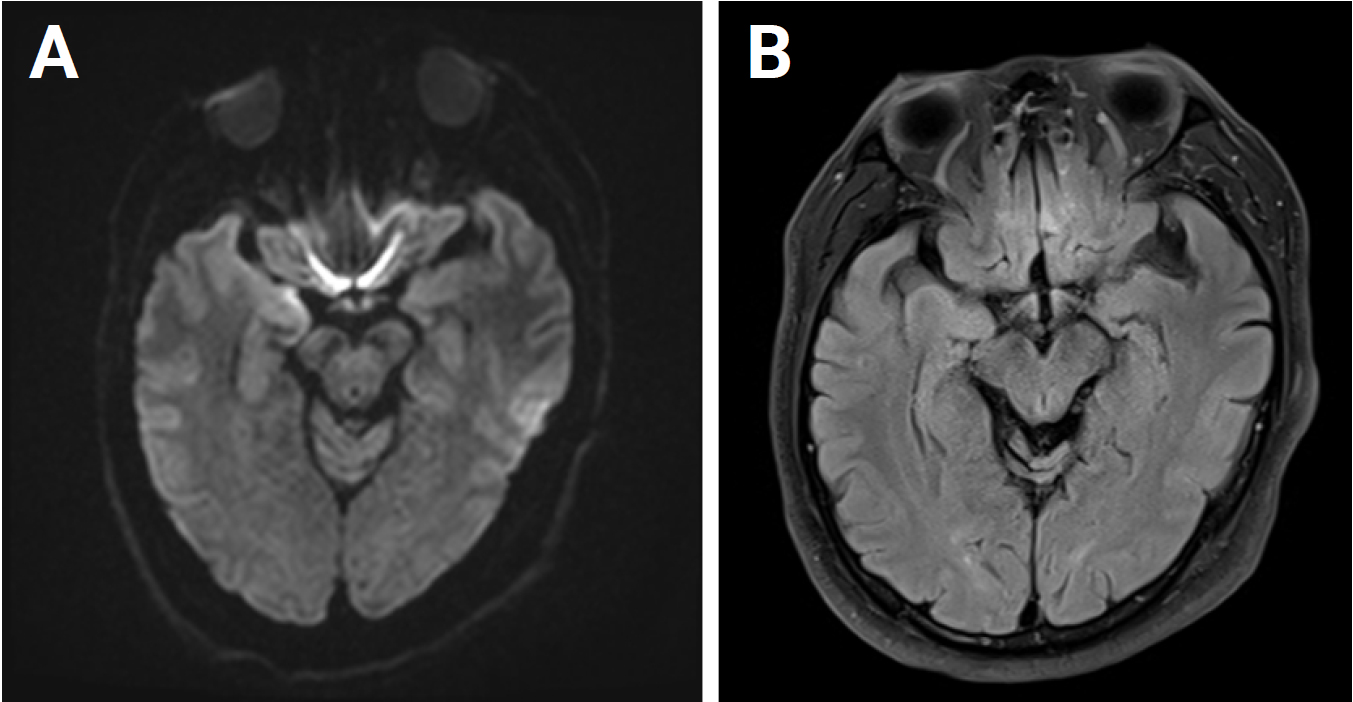
Magnetic resonance imaging obtained on the first day of admission
A diffusion-weighted image (A) and a fluid-attenuated inversion recovery image (B) show no abnormality.
On the first night of admission, the patient developed confusion and fluctuating cognition. He was irritable, hostile toward the nurses in the ICU, and intermittently unaware of his whereabouts. He was suspected to have delirium or postictal confusion and was sedated with intravenous lorazepam. However, the confusion persisted for the next 2 consecutive days. On the third day of admission, EEG revealed sharp and slow waves, followed by rhythmic delta activity. This pattern occurred initially in the right frontotemporal region and subsequently propagated to involve the left frontotemporal area, indicating spatiotemporal evolution (Figure 2). Based on a diagnosis of nonconvulsive status epilepticus with autoimmune encephalitis, intravenous methylprednisolone (500 mg per day) and fosphenytoin (a loading dosage of 25 mg/kg and then a maintenance dosage of 6 mg/kg) were additionally administered. On the fourth day of admission, intermittent high fever (37.8–38.0 °C) developed. Follow-up MRI was performed on the sixth day of admission and revealed diffusion restriction in the right temporal lobe and insular cortex (Figure 3). Immunoglobulin G and polymerase chain reaction (PCR) tests for HSV type 1 in the serum and CSF were positive on the same day. He and his family refused a follow-up CSF examination. Autoimmune antibodies for limbic encephalitis and anti-thyroid antibodies in the serum and CSF were not detected and were not significantly elevated, respectively.
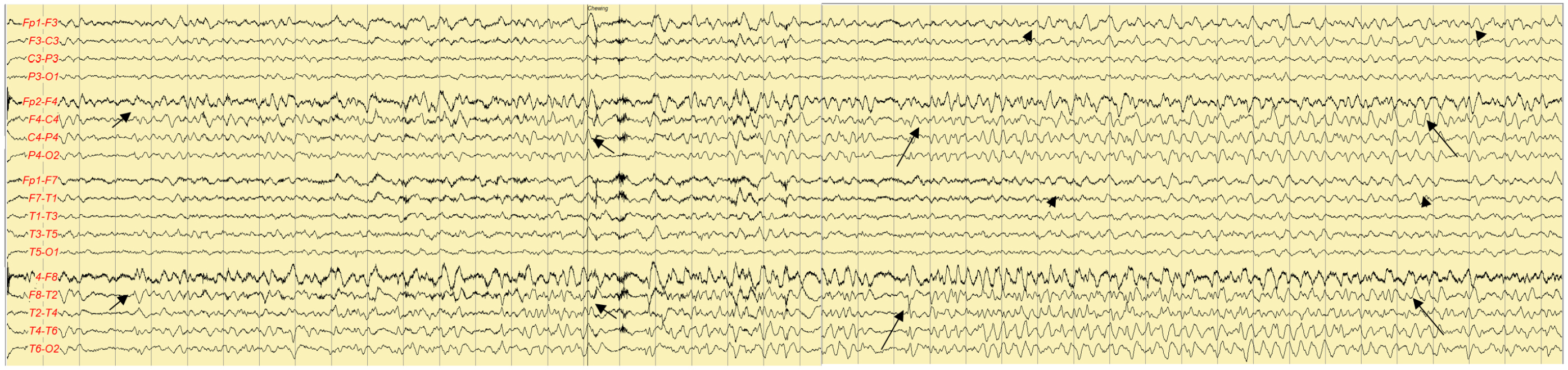
Electroencephalography on the third day of admission
High-voltage, rhythmic sharp and theta waves (long arrows) were followed by rhythmic delta activity (short arrows), which indicates decreasing frequencies and increasing amplitudes, in the right frontotemporal area. The rhythmic activity is spreading to the left frontotemporal area (arrowheads).

Diffusion-weighted images on the sixth day of admission
It shows multiple diffusion restrictions (arrows) in the right temporal area (A) and insular cortex (B).
Methylprednisolone, which was administered from the third day to the sixth day of admission, was discontinued immediately after the HSE diagnosis, and the patient was treated with acyclovir (10 mg/kg three times a day) for 10 days. His K-MMSE score was 29 (–1 in memory recall) at 1 week after the initiation of this new treatment. On the eighth day of admission, lacosamide at a dosage of 50 mg twice daily was prescribed. Subsequently, on the 11th day of admission, fosphenytoin was discontinued, and the dosage of lacosamide was increased to 100 mg twice daily. The patient was discharged without any sequelae. He has been seizure-free for 3 years with valproic acid (500 mg twice a day) and lacosamide (100 mg twice a day). A follow-up EEG performed 18 months after admission revealed no abnormalities.
Written informed consent was obtained for publication of this case report and accompanying images.
Discussion
HSE is a common disease entity and is the cause of approximately 20% of cases of viral encephalitis [5]. To diagnose HSE, neuroimaging, EEG, and CSF examinations are required [4]. In terms of clinical findings, fever and neck stiffness provide clues to the diagnosis; however, they are nonspecific indicators.
Normocellular CSF findings have been reported in several studies on HSE [6,7]. However, in these patients, fever was present in nearly all cases. Without fever and CSF pleocytosis, it is difficult to diagnose HSE, and early diagnosis of this condition is crucial. PCR tests are both sensitive and specific for diagnosing HSE [2], and rapid PCR kits are useful for early diagnosis.
The reason for normocellular CSF in HSE patients is unclear. CSF viral loads were reported to be higher in the pleocytosis group in a previous study [2], but a patient in the normocellular group in the same study showed a high viral load. Genetic factors may also play a role in this mechanism [2]. MRI lesions may sometimes be related to a high viral load, but this is not always the case [2].
Even in elderly and immunocompromised patients, fever is usually present in HSE [6,8,9]. Afebrile HSE is rare [2,8]. Our case did not exhibit any immunological abnormalities and had not taken any medication that could affect the immunological system. The absence of fever may be related to his specific genetically determined immunological traits.
Patients with autoimmune limbic encephalitis (ALE) may present with similar symptoms to those with HSE [10]. As in our case, acute alteration of mentation in the absence of fever and CSF pleocytosis may lead to a misdiagnosis of ALE. We initially misdiagnosed our patient with ALE and administered a high dose of intravenous steroids, which may increase viral loads and lead to more typical clinical and neuroimaging manifestations. Fortunately, our patient promptly ceased methylprednisolone after the correct diagnosis and was administered acyclovir, which resulted in a complete recovery from the illness.
The EEG findings in the acute stage of HSE are varied, including unilateral or bilateral periodic discharges, focal or diffuse slow waves, epileptiform discharges, or seizure activity. However, no specific patterns of EEG are pathognomonic for HSE [11]. Moreover, these findings can also be present in the EEG of ALE patients [12]. Without clinical, laboratory, and neuroradiologic assistance, an EEG alone cannot confirm the diagnosis of HSE.
Brain MRI is the most specific diagnostic modality in HSE. The most pathognomonic lesion is observed in the medial temporal lobes but can also be found in other areas, including the insular, cingulate, and frontobasal cortices [13]. These MRI lesions can also be present in ALE [14], but they are more pathognomonic in HSE.
Diagnosing HSE in patients presenting without fever or CSF pleocytosis and with typical neuroimaging findings is challenging. Misdiagnosing patients with autoimmune encephalitis can lead to immunosuppressive treatment, which may increase viral loads and worsen the clinical condition. Before initiating immunosuppressive treatment, close observation and follow-up neuroimaging or CSF analysis are necessary.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Author Contributions
Conceptualization, Formal analysis, Resources, Supervision:Lee JJ; Writing–original draft: Na Y, Lee JJ; Writing–review and editing: Lee JJ, Kim BK, Lee WW, Kim YS, Yoo I