Introduction
Autoimmune encephalitis is a chronic subacute immune-mediated syndrome with symptoms such as memory impairment, behavior abnormality, psychosis, and seizures [1]. Seronegative autoimmune encephalitis is defined by the absence of detectable autoantibodies in serum or cerebrospinal fluid [2]. Optic neuritis is an inflammatory demyelinating disorder that primarily affects the optic nerve [3].
To the best of our knowledge, there have not been any reported cases of optic neuritis accompanying seronegative autoimmune encephalitis in Korea, and herein we describe a very rare case. This study was approved by the Institutional Review Board of Kyungpook National University Chilgok Hospital (No. 2023-01-010) and written informed consent was obtained for publication of this case
report and accompanying images.
Case report
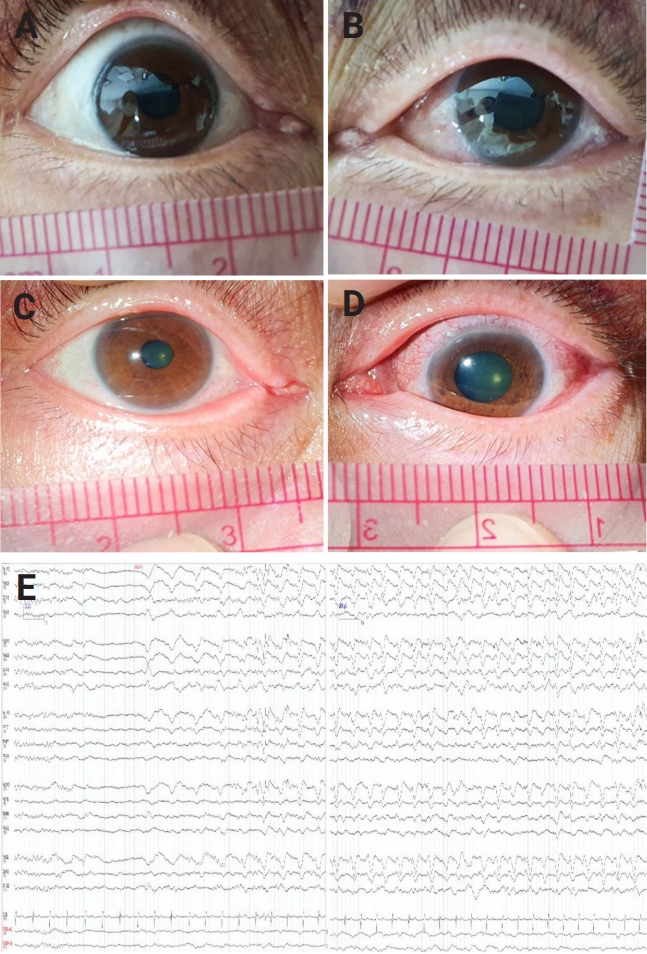
A 65-year-old woman with bipolar disorder visited the neurologic department due to a progressive altered mentality. One month before coming to our hospital, she experienced headache, fever, and fluctuating language disturbance and was admitted to a psychiatric hospital where she was treated previously. However, her speech and cognition gradually deteriorated over the next month, with a progressively drowsier mentality. When she was transferred to our hospital, her vital signs were within normal range without fever. On neurological examination, drowsy mentality and global aphasia were observed, as was anisocoria; the right pupil exhibited a size of 3 mm with normal pupillary light reflex, while the left pupil was dilated to 6 mm in size and direct reflex was absent, while indirect light reflex was preserved. She was observed to have a left relative afferent pupillary defect (RAPD), but no vestibulo-ocular reflex defect. On consultation with an ophthalmologist, funduscopic and slit lamp examinations were performed bilaterally after mydriasis, which revealed no significant findings in the cornea, conjunctiva, or retina, including papilledema or other abnormalities in the retina. However, RAPD was still present (Figure 1). Muscle strength was grade 4 in the bilateral extremities, and the muscle tone of all four limbs was normal. No abnormal hyperactivity of the deep tendon reflexes was observed, and neck stiffness was also not observed.
Magnetic resonance imaging of the brain was performed immediately, but there were no definite abnormal findings including optic nerve sheath enhancement. Electroencephalography (EEG) at the initial presentation showed an evolving 1.5- to 2-Hz generalized rhythmic delta activity superimposed sharp wave, with partial reversibility after intravenous lorazepam (Figure 1). She was diagnosed with nonconvulsive status epilepticus. During video-EEG monitoring, electroencephalographic seizures stopped after administration of levetiracetam (3,000 mg), valproic acid (3,000 mg), lacosamide (400 mg), and perampanel (8 mg).
The cerebrospinal fluid (CSF) study showed normal intracranial pressure, and pleocytosis/protein increases and viral, bacterial, and fungal infections were all excluded based on the CSF profile. The autoimmune work-up was negative for anti-nuclear and anti-neutrophilic cytoplasmic antibodies as well as rheumatoid factor. The results for the following autoimmune and paraneoplastic encephalitis antibodies were also all negative: serum and CSF synaptic antibodies, which included N-methyl-ß┤ģ-aspartate (NMDA), ╬▒-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), dipeptidyl-peptidase-like protein 6 (DPPX), leucine-rich glioma-inactivated 1 (LGI1), contactin-associated protein-like 2 (CASPR2), and gamma-aminobutyric acid type B receptor (GABA-B); serum antiganglioside antibodies; serum paraneoplastic antibodies, which included Hu, Ri, YO, amphiphysin, CV2, PNMA2 (Ma2/Ta), recoverin, SOX1, titin, and anti-myelin oligodendrocyte (MOG) antibody; and CSF oligoclonal band. Laboratory findings including culture and polymerase chain reaction were also all negative. Chest and abdominal computed tomography scans were performed to look for hidden cancers, but none were found.
Finally, she was diagnosed with seronegative autoimmune encephalitis and treated with 1,000 mg of methylprednisolone intravenously (IV) and 0.4 g/kg of intravenous immunoglobulin (IVIG) for 5 days, as well as rituximab (IV, 375 mg/m2, once weekly for 8 doses). She gradually recovered from global aphasia and cognitive impairment, becoming capable of independent daily living. However, optic disc and retinal atrophy were confirmed in the left eye on an ophthalmologist examination 3 months later. During a follow-up period of more than 1 year and 6 months, there was no recurrence of symptoms, and she lived independently without any issues, except for persistent left-sided blindness.
Discussion
The present case with optic neuritis is a clinical presentation of seronegative autoimmune encephalitis. Seronegative autoimmune encephalitis is autoimmune encephalitis without any identifiable pathogenic antibodies [2].
The authors diagnosed this case as autoimmune encephalitis because it met the diagnostic criteria for possible autoimmune encephalitis [4]. In our case, the patient experienced a subacute onset of altered mental status and psychiatric symptoms, seizures unexplained by a previously known seizure disorder, and there was the reasonable exclusion of alternative causes. Also, mental status recovered and nonconvulsive seizures resolved after steroid pulse therapy, IVIG, and rituximab treatment.
Our case is the first report of optic neuritis occurring in seronegative autoimmune encephalitis in Korea, although a few cases of optic neuritis associated with anti-NMDA receptor (NMDAR) antibody encephalitis have been reported worldwide [5,6]. Recently, a few case reports showed encephalitis associated with NMDAR antibodies could mimic neuromyelitis optica involving optic neuritis and transverse myelitis [7], and steroid pulse therapy and plasma exchange were effective for the treatment of optic neuritis with anti-NMDAR encephalitis [8]. According to these reports, optic neuritis resolved with the improvement of autoimmune encephalitis after immunotherapy; however, in our case, despite dramatic improvement of encephalitis, optic neuritis persisted. This difference in treatment results may be due to the low response to immunotherapy of seronegative autoimmune encephalitis compared to autoimmune encephalitis in which antibodies are found. In this case, it is possible that damage to the optic nerve became permanent due to the delay in treatment related to psychiatric hospitalization.
In conclusion, optic neuritis with autoimmune encephalitis is very rare but can occur. It is still unclear if optic neuritis is part of the disease progression of autoimmune encephalitis; some theorize that the receptor of autoantibodies is expressed in developing oligodendrocyte processes and may mediate the regeneration of retinal ganglion cells. This case report demonstrates that in rare cases of optic neuritis associated with seronegative encephalitis, timely immunomodulatory treatment may be critical to preserving vision.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print